
英文标题:Haplotype‐resolved genome of a papeda provides insights into the geographical origin and evolution of Citrus
发表期刊:Journal of Integrative Plant Biology
影响因子:9.3
合作单位:西南大学柑桔研究所等
百迈客生物为该研究提供了基因组测序及组装等相关工作。
研究背景
柑橘是世界上种植最广泛的水果之一,目前在140多个国家和地区种植。虽然过去的二十年里柑橘植物的种群结构和遗传多样性已被调查,但由于其丰富的资源类型和杂交频繁,许多柑橘物种的起源和进化仍不清楚。野生柑橘物种和原始柑橘保存了丰富的遗传资源和遗传多样性,可用于阐明柑橘物种的起源和进化。2015年,在西双版纳发现一棵200多年的野生柑橘树大翼叶柑橘(DYC002),被认为是研究柑橘进化及苦味驯化的很好材料。
研究方法
DYC002单倍型基因组组装:PacBio HiFi 180×;llumina 75×;HIC 134×
群体进化研究:378份具有代表性的材料进行全基因组重测序分析;平均测序深度32.6×
DYC002转录组测序:幼叶、茎、花、小果和根(3个生物学重复)
研究结果
作者利用53.7G PacBio HiFi、22.6G illumina reads和40.1G Hi-Creads,首先组装出318.71 Mbp大小的contig版本基因组,contig N50为2.48 M,接着利用HIC数据将contig 挂载到染色体上,得到得到294 Mbp的基因组,BUSCO评估完整性大于98%,LTR组装指数(LAI)得分高达21.1,测序数据回比率为97.43%,进一步支持了组装的完整性。最后对DYC002基因组注释,共注释到34,652个蛋白编码基因和215,693个重复序列。重复序列多为LTR,尤其是Gypsy LTR和Copia LTR,占组装基因组的23.62%。比较基因组发现DYC002有191个基因家族正在快速进化。GO富集分析表明,这些快速进化的基因主要富集于萜烯合酶活性、倍半萜合酶活性、(4S)-柠檬烯合酶活性、(E)-柠檬烯合酶活性、葡萄糖苷酶活性和-葡萄糖苷酶活性等功能,表明DYC002在进化过程中香气和风味发生了显著变化。
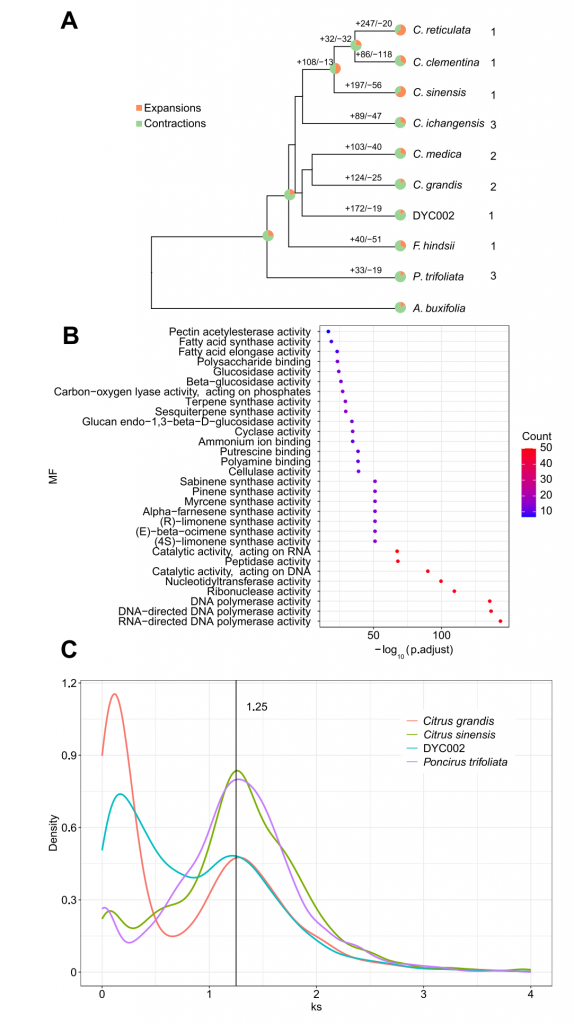
图1-比较基因组分析
利用CCS数据和HIC数据,作者组装出DYC002的两套单倍型基因组,长度分别为294和300 Mbp,注释的基因数量分别为28000和27785个;BUSCO完整度分别为97.39%和97.33%。通过比较分析,两单倍型基因组间存在175万个SNPs,303,686个Indels,转录组分析表明,两个单倍型中等位基因的表达高度一致。叶片中等位基因特异性表达基因(ASEGs)数量最高,而花中数量最低。在单倍型1和单倍型2的组装中分别发现了535个和499个单倍型特异性基因。
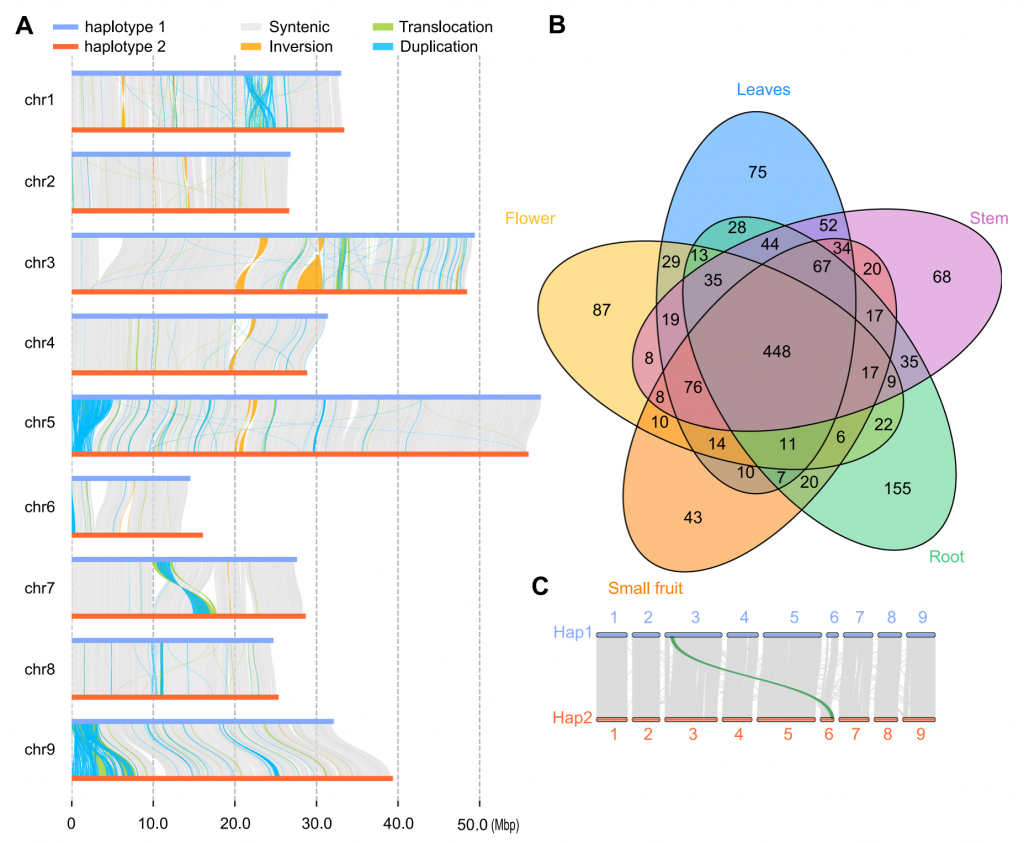
图2-单倍型基因组比较分析
为了说明柑橘物种的进化历史,作者从378份具有代表性的柑橘材料中获得了基因组重测序数据,利用核基因组SNPs进行的系统发育分析,除了莽山野柑,柑橘属可分为两个进化支,一支以宽皮柑橘、宜昌橙、香橙、甜橙等为代表,另一支包括大翼橙、澳指檬、澳沙檬、枸橼、红河大翼橙、柚、酸橙等。推测柑橘属植物从起源中心朝东南亚和中国东部两个方向进行演化和传播。选择性清除分析表明,苦味在柑橘驯化过程中具有较高的选择,共鉴定出672个选择性扫描区,一些参与类柠檬苦素和类黄酮生物合成的4-香豆酸-CoA连接酶、糖苷转移酶受到选择。
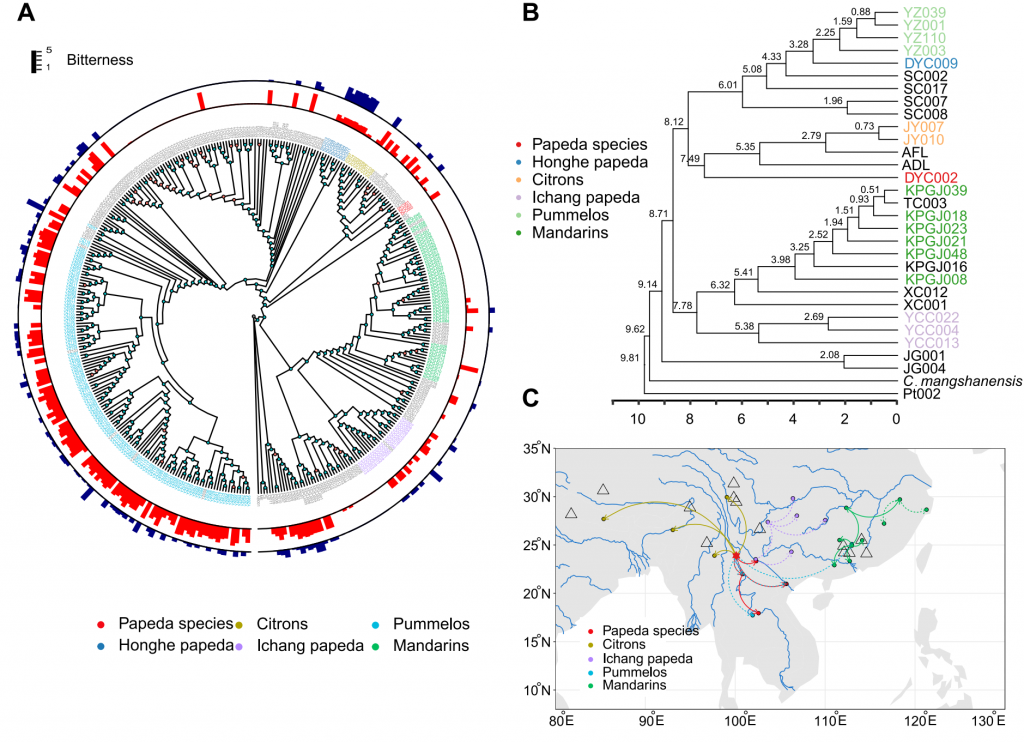
图3-群体进化分析
研究总结
高质量基因组的发表对了解柑橘发挥重要作用。然而,由于其复杂的遗传背景,许多柑橘物种的起源和进化仍不清楚。作者从头组装了DYC002的294mbp染色体水平的基因组,对两个单倍型基因组比较,发现了1.2%的基因组内变异,包括175 万个SNPs,149,471个插入和154,215个缺失。以该基因组为参考,对378份具有代表性的柑橘材料进行了重测序和系统发育分析,作者研究柑橘属可分为两个进化支,证实了柑橘核心种的主要起源中心是华南地区,特别是喜马拉雅-横断山脉。该研究为了解柑橘品种的起源和进化提供了新的视角,并可能为柑橘育种和改良提供有价值的基因组资源。
]]>

文章题目:T2T?genome,?pan-genome?analysis?and?heat?stress response?genes?in?Rhododendron?species
发表期刊:iMeta
影响因子:23.8
合作单位:贵州大学,华北理工大学
百迈客生物为此项研究提供基因组测序及部分分析服务。
研究亮点:
① 该研究首次报道了具有13条染色体的高质量端粒到端粒(T2T)的百合杜鹃基因组;
② 基于15个杜鹃属植物基因组,对杜鹃属植物进行了泛基因组分析;
③ 结合基因组测序和全转录组测序,鉴定了几个与热胁迫相关的关键基因和miRNA,为杜鹃属植物的比较基因组学和功能基因组学研究提供了丰富的资源。
研究背景
杜鹃花属于杜鹃花科,是木本植物中最大的属之一。全世界大约有1000种杜鹃花,中国是重要的分布中心。它们在喜马拉雅-横断山脉经历了进化辐射,横断山脉是世界生物多样性的热点。杜鹃花属植物因其观赏价值而在园艺中占有重要地位。全球气候变化导致温度升高,而热胁迫会影响植物的生长发育。然而,杜鹃花植物通常适应较冷的气候。利用多组学分析和分子生物学技术研究杜鹃花对高温胁迫的响应机制,对选育耐热品种、扩大杜鹃花栽培范围具有重要意义。
材料及方法
T2T基因组组装:Rhododendron liliiflorum
泛基因组分析:4个本次组装的基因组+11个已报到的杜鹃属基因组
全转录组:CK热处理、热处理3天(H3)和6天(H6)条件下进行了全转录组测序
两对具有代表性的miRNAs和相关靶基因进行功能验证。
研究结果
1.杜鹃属植物基因组测序、组装与评价
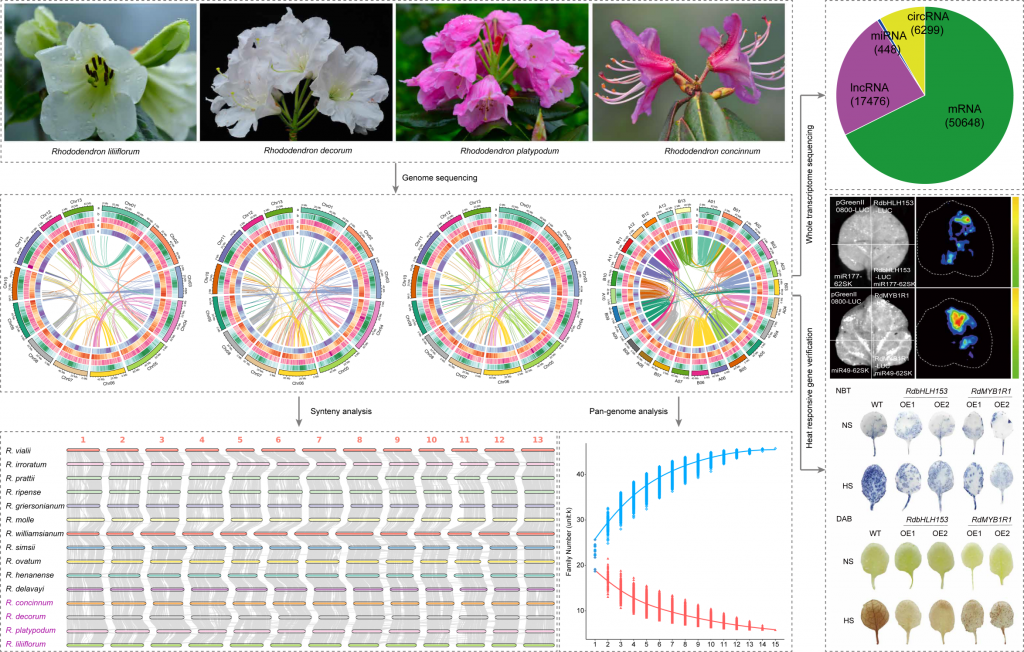
该研究通过PacBio HiFi、Oxford Nanopore Technology(ONT)、Illumina和Hi-C技术(图1A,表S1-6)对四种杜鹃花植物(Rhododendron liliiflorum、Rhododendron decorum、Rhododendron platypodum和Rhododendron concinnum)进行从头基因组测序。通过K-mer估算的R. liliiflorum、R. decorum、R. platypodum和R. concinnum的基因组大小分别为759.08 Mb、581.05 Mb、593.47 Mb和1356.22 Mb,并通过流式细胞术进一步验证(表S1,图1B)。
作者发现,R. concinnum的基因组几乎是其他三个物种的两倍大。因此,作者们利用流式细胞术进一步分析了染色体核型,首次发现R. concinnum为四倍体,核型为2n=4x=52,与其他三个二倍体物种(2n=2x=26)有明显差异(图1C,表S1)。
4个物种的基因组大小分别为793.25 Mb、649.87 Mb、652.27 Mb和1321.11 Mb(表S1)。经Hi-C检测,4个种的染色体锚定率均在97.90%以上(图1D,表S5)。作者获得了4个高质量的组装基因组,支架N50大于48.68 Mb。核心真核基因作图法(CEGMA)值从95.63%到99.56%,基准通用单拷贝同源序列(BUSCO)值从96.65%到97.34%,读取作图率超过99.40%(表S7)。
最重要的是,作者获得了一个高质量的R. liliiflorumT2T基因组,该基因组由13条染色体组成,检测到24个端粒和13个着丝粒(图S1A,表S8-11)。其中11条染色体在端粒与端粒之间没有间隙,另外2条染色体只有一个间隙。百合杜鹃花基因组的重叠群N50大于58.56MB,大于以往大多数杜鹃基因组的重叠群N50。采用BUSCO软件对基因组完整性进行评估(96.65%),基因组一致性质量值(QV)为43.71(表S1)。基因组LTR组装指数(LAI)值为21.15(图S1B),表明已达到最高质量水平(LAI≥20)。
重复序列占4个基因组的49.10%以上,以长末端重复序列(LTRs)最多(图1E,表S11)。在这四个基因组中,共预测到41406、41084、40556和83203个基因(表S12)。检测到超过97.15%的BUSCO基因,说明预测的完整性较高(表S13)。NR、eggNOG、GO、KEGG、TrEMBL、KOG、Swissprot和Pfam数据库对92.16%以上的基因进行了注释(表S14)。共检测到2355、4862、2852和9511个非编码RNA(表S15)。
2.15个杜鹃属植物泛基因组分析
杜鹃属植物以其多样的花朵展示而闻名,近年来,随着第一个R. delavayi基因组的公布,一些基因组被解码,引起了科学界的高度关注。报道了几种杜鹃属植物的基因组,如R. griersonianum、R. Henanense、R. Irroratum、R. kiyosumense、R. Ripense、R. Vialii、R. nivale和R. williamsianum。这些基因组为泛基因组研究奠定了基础。基于这四个高质量基因组以及11个先前发表的基因组,对杜鹃属植物进行了泛基因组分析(图1F,表S16)。选择T2T水平的R. liliiflorum基因组作为参考。该泛基因组通过添加394.57?Mb和14424个基因,扩展了T2T水平的R. liliiflorum基因组。15个物种的基因家族数量为45731个,包括5734个核心基因家族、37027个可有可无的基因家族和2970个私有基因家族(图1G、图S2A、表S17)。利用打乱图分析了15个物种间基因家族共享与唯一性的关系。最后,作者基于聚类分析构建了基因家族存在与否的分布图(图1H)。在2970个私有基因家族中,R.irroratum(1705)的物种特异性基因最多(表S17,图S2B)。功能富集分析表明,“倍半萜和三萜生物合成”和“亚油酸代谢”途径显著富集(图S3)。共鉴定出121185个核心基因,R. ovatum基因组的数量最多(9847)(图S2B)。功能富集分析表明,与花的颜色和香味相关的基因途径显著富集,如柠檬烯和蒎烯降解(图S4)。
3.15个杜鹃属植物基因组的变异分析
基于以T2T基因组为参考的泛基因组分析,作者对杜鹃花的单核苷酸多态性(SNPs)、插入和缺失(InDels)以及结构变异(SVs)等变异进行了全面鉴定(图1I-L,图S5)。四倍体R. concinnum具有最多的SNP(1876446)和InDels(447281)(图1I,表S18-19)。功能富集分析表明,含有SNPs和InDels的基因在“碳代谢”和“氨基酸生物合成”途径中显著富集。R. concinnum的SV数量最多,达到7694(表S20)。同时,作者进一步将SVs细分为重复(DUP)、易位(TRANS)和反转(INV),发现在大多数杜鹃属植物中,前者的数量超过后者(图S6-7)。SVs基因与SNPs或InDels基因相比表现出明显的模式,主要集中在RNA聚合酶和mRNA监控途径上。
4.15个杜鹃花基因组的LTR分析
该研究在15个杜鹃属物种的全基因组中鉴定了70759个LTR,其中R. griersonianum基因组的LTR数量最多(7323)(表S21)。作者发现,在过去的一百万年中,大多数杜鹃花物种只经历了一次插入事件的爆发,而R. delavayi、R. molle和R. williamsianum经历了两次插入事件的爆发。两个事件分别发生在1.53mya和2.94mya。作者对15个物种的LTR进行聚类,得到每个聚类中的共享LTR。结果表明,共有2622个LTR聚类,其中R. platypodum最多(531个)。R. liliiflorum中特异性LTRs数量最多(109个),而R. williamsianum中未发现物种特异性LTRs。聚类图显示,R.williamsianum与其他物种共享LTR的比例最高(图1M)。此外,LTR在染色体中部的分布密度大于两端(图1N)。
5.15个杜鹃属植物基因组的共线性分析
通过共线性分析,研究发现15个杜鹃基因组普遍表现出良好的共线性(图1O)。共线性区组数从336个(R. henanense?vs?R. delavayi)到692个(R. irroratum?vs?R. prattii)。此外,还发现了一些基因组转座现象,如R. ovatum7号染色体的末端区域与R. simsii和R. henanense。
图 1. 4种杜鹃属及11种已发表杜鹃属植物的基因组分析 (A)四种杜鹃花的开花照片;(B)通过k-mer进行基因组调查。(C)流式细胞术检测染色体核型;(D)基因组组装的Hi-C接触图;(E)转座子、SSR、基因密度和GC含量在染色体上的分布;(F)核心(红色)和非核心(蓝色)基因家族数量趋势图;(G)核心集群、可有可无集群和私有集群的家族数量;(H)核心、可有可无和私有集群的存在和缺失分析;(I)百合杜鹃花基因组中基因密度、SNP和InDel变异的分布情况;(J-L)以T2T基因组为参照,研究了端粒红豆杉、鸭嘴兽和短柄红豆杉基因组的同源性和重排;(M)两个物种之间共享LTR的比例(Rconc:?R. concinnum; Rdeco:?R. decorum; Rdela:?R. delavayi; Rgrie:?R. griersonianum; Rhena:?R. henanense; Rirro:?R. irroratum; Rlili:?R. liliiflorum; Rmoll:?R. molle; Rovat:?R. ovatum; Rplat:?R. platypodum; Rprat:?R. prattii; Rripe:?R. ripense; Rsims:?R. simsii; Rvial:?R. vialii; Rwill:?R. williamsianum);(N)LTR在染色体上的分布。x轴代表染色体位置的百分比,y轴代表LTR的插入时间;(O)15种杜鹃属植物的基因组共线性。右边的数字表示共线块编号。
6.热反应基因的全转录组测序与检测
为了探索杜鹃花的耐热基因和调控机制,该研究在CK热处理、3天热处理(H3)和6天热处理(H6)条件下进行了全转录组测序(图2A,表S22)。共鉴定出50648个mRNAs、17476个lncRNAs、448个miRNAs和6299个circRNAs(图2B)。此外,在CK、H3和H6处理中,632个mRNAs、21个lncRNAs和6个miRNAs的表达和共享存在差异(图2C)。
7.候选基因的功能验证
该研究选择了两对具有代表性的miRNAs和相关靶基因进行功能验证。热处理后3h和6h,靶基因表达显著上调,小RNA表达显著下调。作者进一步研究了miR177对RdbHLH153(Rhdel02G0118700)表达和miR49对RdMYB1R1(Rhdel08G0208700)表达的影响。将萤火虫荧光素酶分别融合到RdbHLH153和RdMYB1R1的C端,并将miR49和miR177分别插入SK载体(图2D)。结果显示,RdbHLH153中的miR177和RdMYB1R1中的miR49的靶位点略有改变(图2E)。用RdbHLH153/RdMYB1R1与空SK载体(混合并渗透)或RdbHLH153与miR177(RdMYB1R1和miR49)的混合物对烟草单叶渗透区进行渗透。均显示荧光素酶信号的诱导,而R. delavayi中过表达的miR177和miR49可消除RdbHLH153/RdMYB1R1产生的信号(图2E-G)。
为了进一步研究RdbHLH153和RdMYB1R1在热胁迫中的作用,采用花浸法获得了过表达RdbHLH153和RdMYB1R1的转基因拟南芥株系(表S23)。36 h热处理后,转基因植株的生长明显优于WT(图2H)。经过热处理后,幼苗恢复正常状态5天,发现转基因植株转活,WT叶片全部变黄。说明RdbHLH153和RdMYB1R1在提高耐热性中起重要作用。DAB和NBT染色显示,与野生型植株相比,RdbHLH153/RdMYB1R1 OE株系中H2O2和O?2的含量显著降低(图2I)。
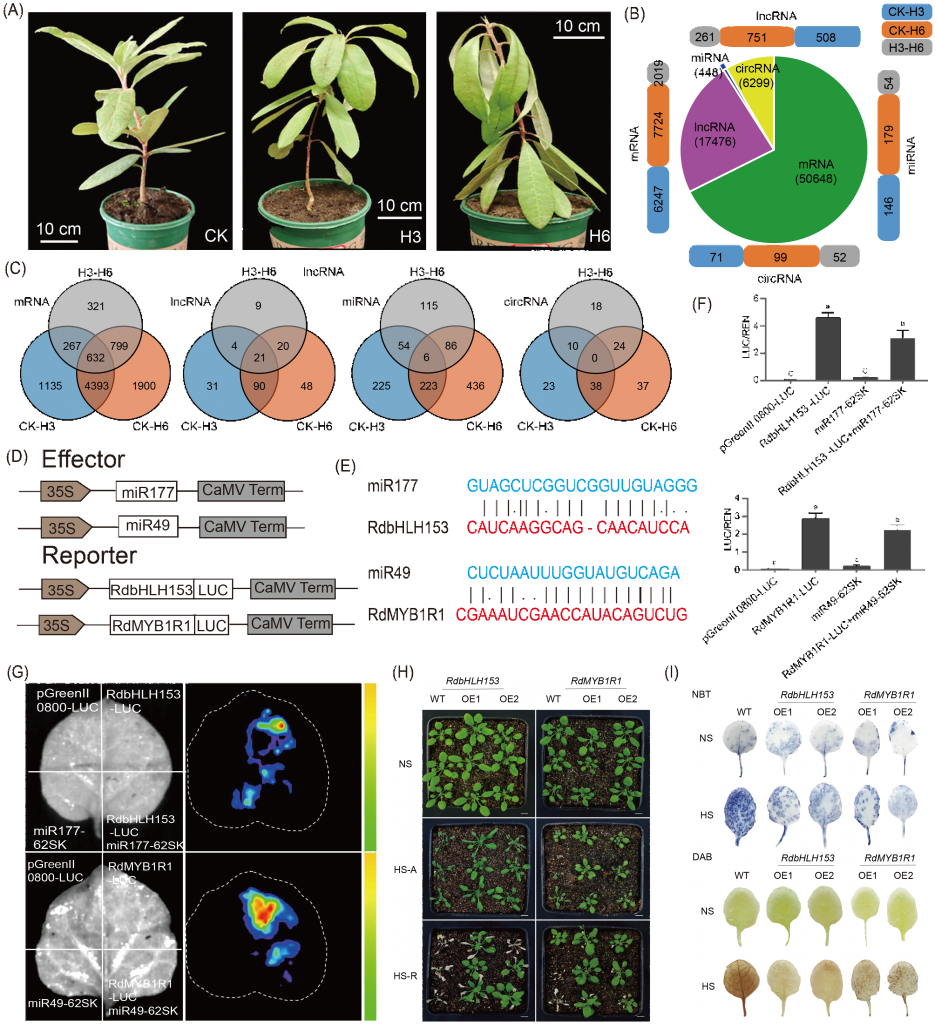
图2. miRNA和靶基因的全转录组分析及功能验证 (A)R. delavayi的对照(CK)和热处理(H3和H6);(B)mRNA、lncRNA、miRNA和circRNA的鉴定和差异表达分析;(C)三种比较中特异性和常见的差异表达RNA;(D)效应器和报告器结构图;(E)miR49和miR177靶点突变示意图;(F)荧光素酶测定统计。误差条表示三个重复的SEs(*,p?< 0.05)。(G)荧光素酶成像分析;(H)RdbHLH153和RdMYB1R1的过表达增强了耐热性。NS表示无热应激,HS-A表示36 h热应激,HS-R表示36 h热处理后在常温下恢复5天;(I)二氨基联苯胺(DAB)和硝基蓝四氮唑(NBT)染色法检测转基因株系和WT在无热胁迫(NS)和热胁迫(HS)下的H2O2和O?2。
内容来源于iMeta
]]>
研究背景
葡萄(Vitis)在全球范围种植广泛,即可鲜食又可加工成葡萄酒等,具有很高的经济价值,是农民致富、乡村振兴、人民美好生活中不可或缺的重要园艺作物。葡萄属于葡萄科葡萄属植物,该属包括两个亚属,麝香葡萄亚属(Muscadinia Planch)和真葡萄亚属(Euvitis Planch),共计70余个种;根据地理分布可将其分为三大类群,欧亚种群、北美种群和东亚种群。
葡萄作为最古老的驯化作物之一(约公元前11,000年),漫长的持续驯化和多年的育种改良导致现代葡萄品种的遗传多样性缩小和抗性丢失,使其易受病虫、逆境等各种不利条件的影响。近年北美葡萄泛基因组(Genome biology, 2023),欧洲葡萄泛基因组(Nature Genetics, 2024)的相继报道,野生葡萄以其丰富的遗传多样性和超强的抗性基因再次受到关注,但以大群体染色体级基因组组装为基础,涵盖主要品种,特别是包含东亚野生种葡萄的属级泛基因组一直未见报道。因此构建涵盖整个葡萄属的泛基因组将成为了解葡萄遗传多样性、开展功能基因组学研究、识别隐性遗传特性以及葡萄精准改良的关键资源。
材料方法及研究结果
该研究首先组装了酿酒葡萄(Vitis vinifera)霞多丽的完整单倍型基因组,并首次基于ChIP-seq数据鉴定了葡萄着丝粒序列,解析着丝粒的卫星重复序列结构特征,发现单倍型着丝粒之间的显著差异,表明其快速进化。其次,该研究通过对591个葡萄材料的群体基因组分析,选取了72个代表性葡萄材料(包括25个野生种和47个栽培种)进行染色体水平的单倍型基因组组装(图1),基因组验证表明这些单倍体基因组序列具有较高的质量和完成度。
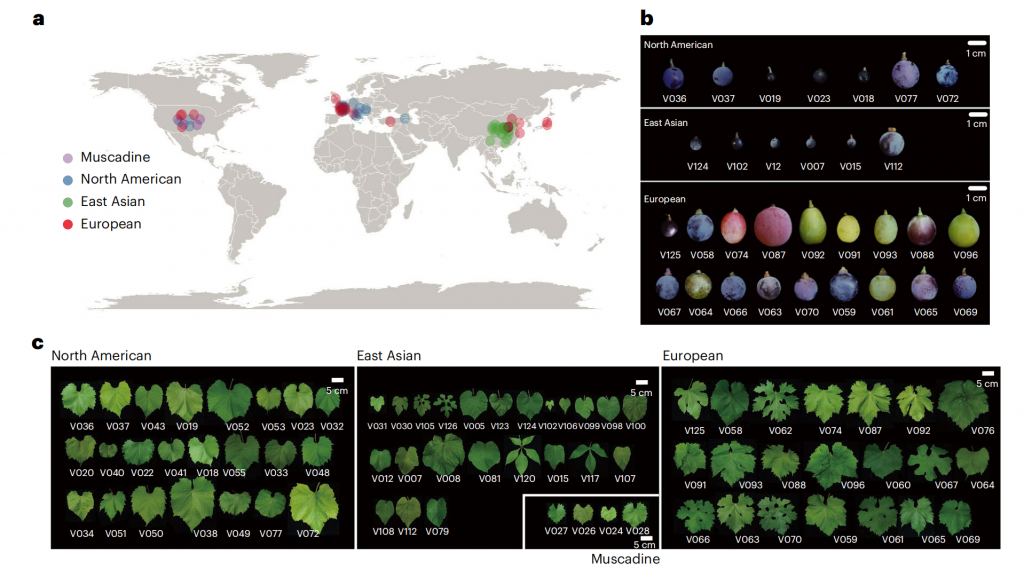
图1-葡萄样品全球地理分布及其果实和叶片形态
鉴于葡萄基因组因频繁杂交而具有的高度杂合性,单倍型基因组不仅能精确解析杂合区域序列,而且对分析葡萄的育种历史也至关重要。该研究构建了单倍型系统发育进化树,揭示了葡萄属植物复杂的育种历史和丰富的遗传多样性。结果显示,北美和欧洲品种存在较多内部杂交,而东亚品种的内部杂交较少,跨大洲杂交事件有限(图2)。特别是东亚葡萄极少被开发的遗传多样性,表明了其潜在的巨大育种价值。此次发布的东亚野生葡萄基因组为葡萄遗传育种研究提供了强有力的资源支持。
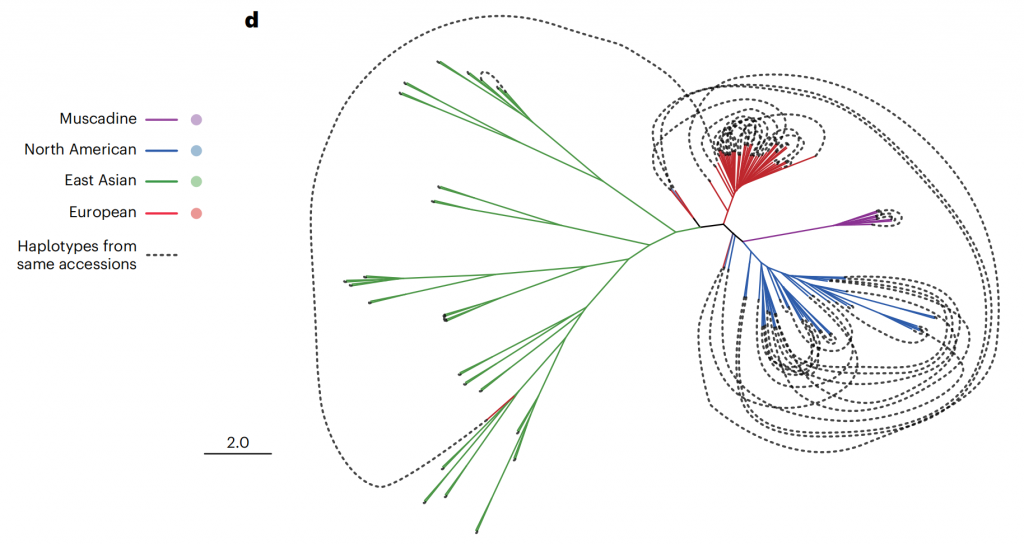
图2-144个单倍型基因组系统发育树
该研究还分析了72份葡萄材料的泛基因组家族,发现了超过6.4万个基因家族,包括不同数量的核心、可变和私有的基因家族。拟合曲线表明,该研究的泛基因家族数量趋向饱和,表明葡萄泛基因组接近闭合。研究还系统分析了葡萄属免疫受体蛋白基因家族NLR,结合三大种群的葡萄和圆叶葡萄抗霜霉病数据发现,TIR-NBARC-LRR家族的NLR基因在抗病(野生葡萄)和感病葡萄(栽培葡萄)中存在显著数量差异,因此有可能是葡萄抗霜霉病表型差异的重要因素之一。该超级泛基因组分析为研究葡萄属的遗传多样性、进化历史及功能基因挖掘提供了全面的基因组基础(图3)。

图3-葡萄属72份代表性材料超级泛基因组基因家族图谱
其次,该研究进一步绘制了目前比较完整的葡萄基因组遗传结构变异(SV) 图谱,助力挖掘与抗性及资源利用效率等重要性状相关的功能基因(图4)。该研究通过全基因组序列比对和变异检测,在67个Euvitis葡萄样本中鉴定出132,518个非冗余结构变异。功能富集分析表明,这些SV与葡萄叶片形态、病原识别及生物刺激感知相关。通过与已知分子标记的比较,该研究鉴定到霜霉病抗性分子标记Rpv3相关SV事件,并发现三大种群中该SV位点的不同单倍型与葡萄霜霉病抗性有显著关联性,证明该SV是一个关键抗病分子标记,突出了超级泛基因组图谱的价值。
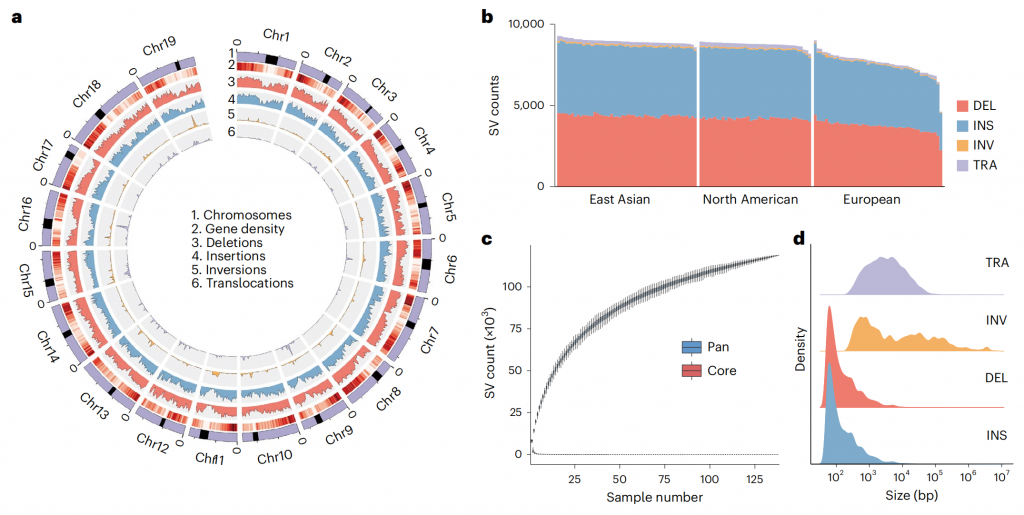
图4-葡萄属72份代表性材料结构变异图谱
最后,该研究使用葡萄结构变异图谱和基于霞多丽完整基因组序列,构建了涵盖欧、亚、美三大种群的图形泛基因组。基于该图形泛基因组,该研究对113个葡萄样本的霜霉病抗性表型、气孔表型以及霜霉病侵染转录组数据进行了基因组变异的eQTL分析(图5),鉴定出63个SV-eQTL和1,808个SNP-eQTL与葡萄抗霜霉病显著相关。在63个与霜霉病抗性密切相关的SV的辅助下,该研究定位到一个氨基酸转运蛋白基因VvLHT8。进一步的分子功能实验发现VvLHT8可能通过负调控葡萄水杨酸合成和气孔免疫反应进而抑制葡萄抗病性,证实了高质量泛基因组辅助的多组学关联分析在作物重要农艺性状分子标记开发及功能基因挖掘中的高效性。该研究构建的葡萄属超级泛基因组不仅加深了对葡萄生物进化和育种改良的理解,也为精准改良葡萄抗病性和多样性提供了重要的科学基础。
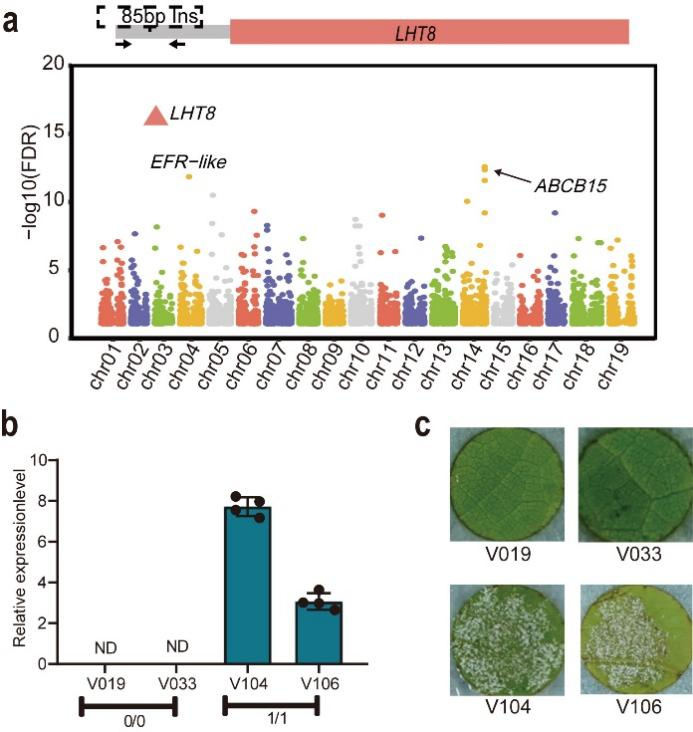
图5-超级泛基因组图谱辅助SV-eQTL鉴定及VvLHT8基因功能验证
研究总结
该研究提供了比较全面的葡萄属基因组资源,有助于全面解析葡萄基因组的复杂性和多样性,从而高效发掘并利用葡萄特别是野生葡萄种中的优异基因。该研究结合葡萄属超级泛基因组、群体转录组学和表型组学,对葡萄属遗传多样性和重要农艺性状形成机制的深入探索,为未来培育超级葡萄提供了理论基础和新的思路(图6),标志着葡萄基因组研究迈进新的阶段,也必将加速推动我国葡萄种质创新和葡萄产业的高质量发展。
图6-葡萄属超级泛基因组构建和应用
以下小编列举三篇成功案例,解析群体在QTL定位中的应用。
当我们针对一群体,关注性状较多时,可参照案例一,整篇文章通过群体图谱构建,QTL定位后几乎没有验证工作,可以帮我们拿到一个群体QTL的基础数据。
案例一

发表期刊:Plant Physiology and Biochemistry
影响因子:6.1
合作单位:江苏省徐淮区徐州农业科学研究所农业农村部甘薯生物学与遗传改良重点实验室
实验方法:Xin24×Yushu10杂交中选择212个F1材料: 特异性位点扩增片段测序(SLAF-seq);遗传图谱构建;数量性状位点(QTL)定位;GWAS关联(F1群体)
表型检测:多年多表型收集
百迈客生物为该研究提供了群体测序及部分数据分析服务。
甘薯作为一个全球重要的作物,其含有丰富的营养成分以及对不同环境的适应能力。然而,由于其自交不亲和,高杂合度等特性,使其遗传特征的研究相对较少。作者从Xin24×Yushu10杂交中选择212个F1材料,SLAF-seq测序,获得亲本26.73×,子代52.25×的测序数据,依据SNP及百迈客生物自主研发的HighMap构图软件,生成一个长度为2441.56 cM、平均图距为0.51 cM的遗传图谱。
基于连锁图谱,鉴定出26个QTL,解释了6.3-10%的表型变异,包括6个最长藤蔓长度 QTL、6个单株产量 QTL、10个干物质含量QTL、1个淀粉含量 QTL、一个可溶性糖含量QTL和2个类胡萝卜素含量QTL。该研究结果对甘薯的标记辅助育种和基因克隆具有重要意义。
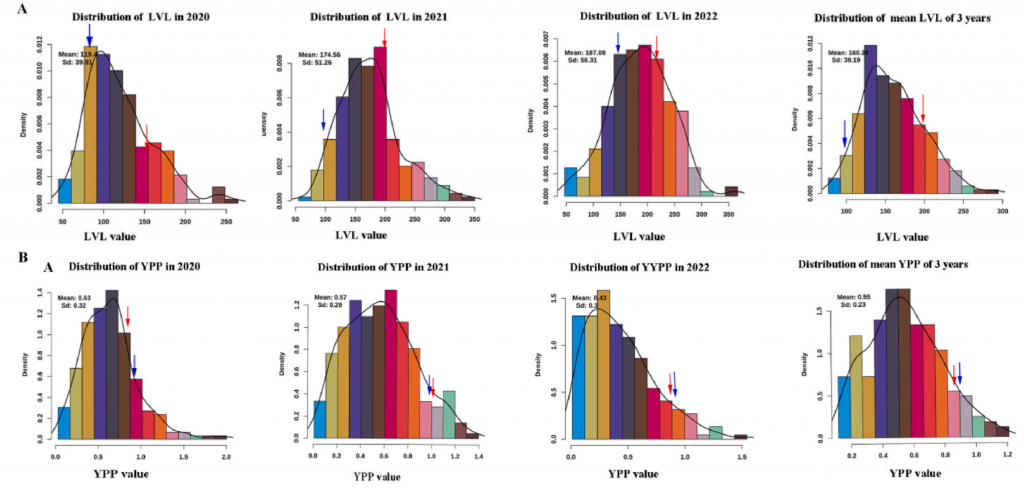
图1-表型检测
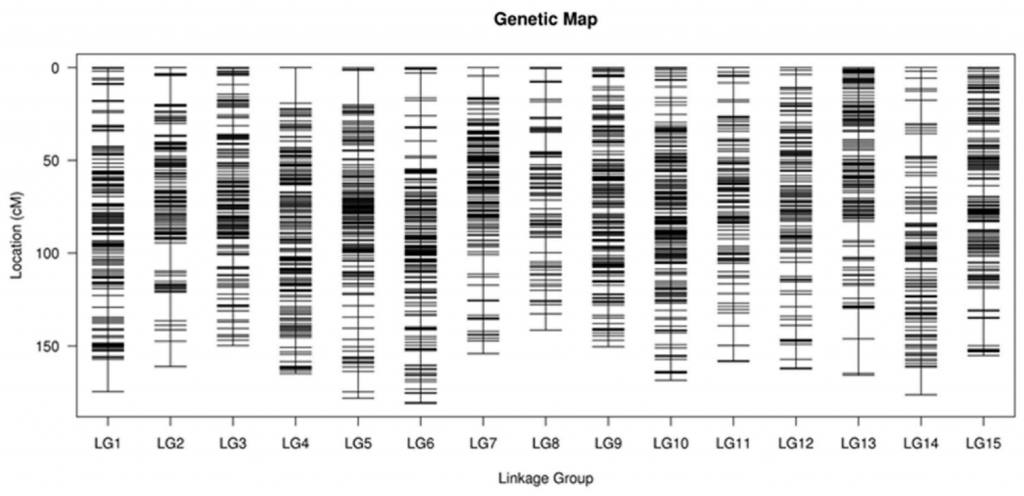
图2-遗传图谱构建
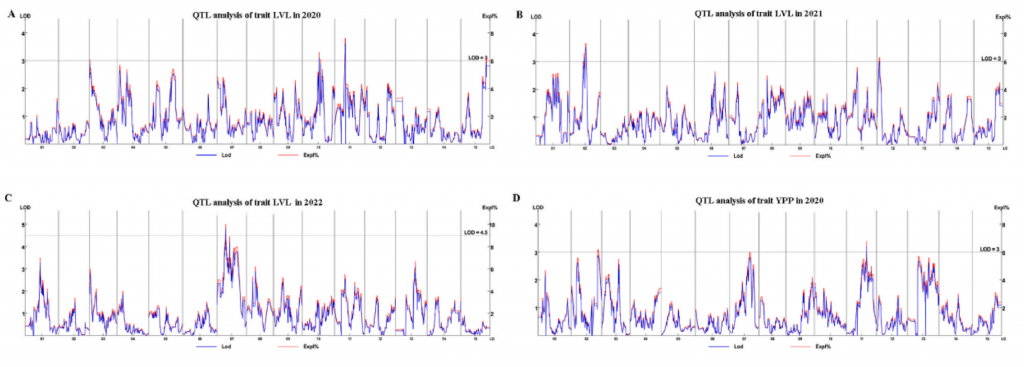
图3-QTL定位
前文是对多个性状的连锁分析,当我们关注单个性状时,BSA无疑是高性价比的初定位选择,当然,这就意味着我们得做到基因的精细定位与克隆,除去传统图位克隆的方式,转录组,蛋白组,自然群体GWAS,基因组都可助力基因克隆,如果想发高水平的文章,基因的功能探索也是必不可少的。
成功案例二

发表期刊:Plant Biotechnology Journal
影响因子:10.1
发表单位:河南农业大学
实验方法:荚果大小/重量差异显著2份Virginia-type花生材料( ND _ L和ND _ S)杂交,产生遗传群体;F2:3群体BSA-seq(20+20混池);F6:7 和F6:8群体精细定位;基因克隆;系统进化分析;亚细胞定位;免疫荧光;酵母双杂交;pull-down;CO-IP;番茄拟南芥转化实验等。
百迈客生物为该研究提供了群体测序及部分数据分析服务。
花生荚果大小是决定花生产量的关键农艺性状,为了鉴定控制花生荚果大小的基因,该研究对188份核心种质进行鉴定,并选取荚果大小/重量差异显著的2份花生材料构建F2群体,BSA-Seq获得了288.58 Gb的原始数据。利用285914个高质量SNPs 和70 759个 InDel,将控制荚果大小的基因定位在07染色体1.17 Mb的区间内。作者从F6:7和F6:8群体中开发了15个多态性标记并对个体进行基因分型,精细定位QTL到KASP 9 和In Del15之间20.4 kb的区间内。该区间仅包含1个预测的非同义突变基因和InDels,将该基因命名为PSW1。
PSW1编码一个LRR – RLK蛋白激酶,等位基因PSW1HapII赋予了PSW1更高的表达水平和对其辅助受体AhBAK1更强的亲和力,以上调PSW1 – based途径,调节花生荚果大小。此外,PSW1HapII的过表达增加了多种植物的种子/果实大小。
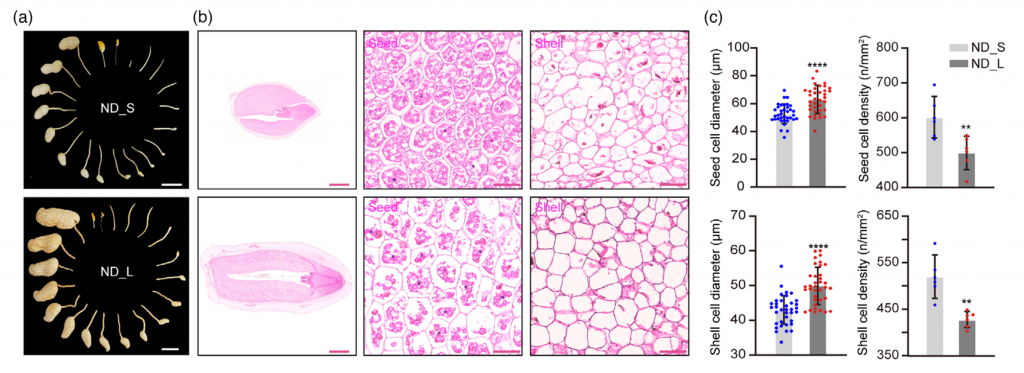
图4-表型检测
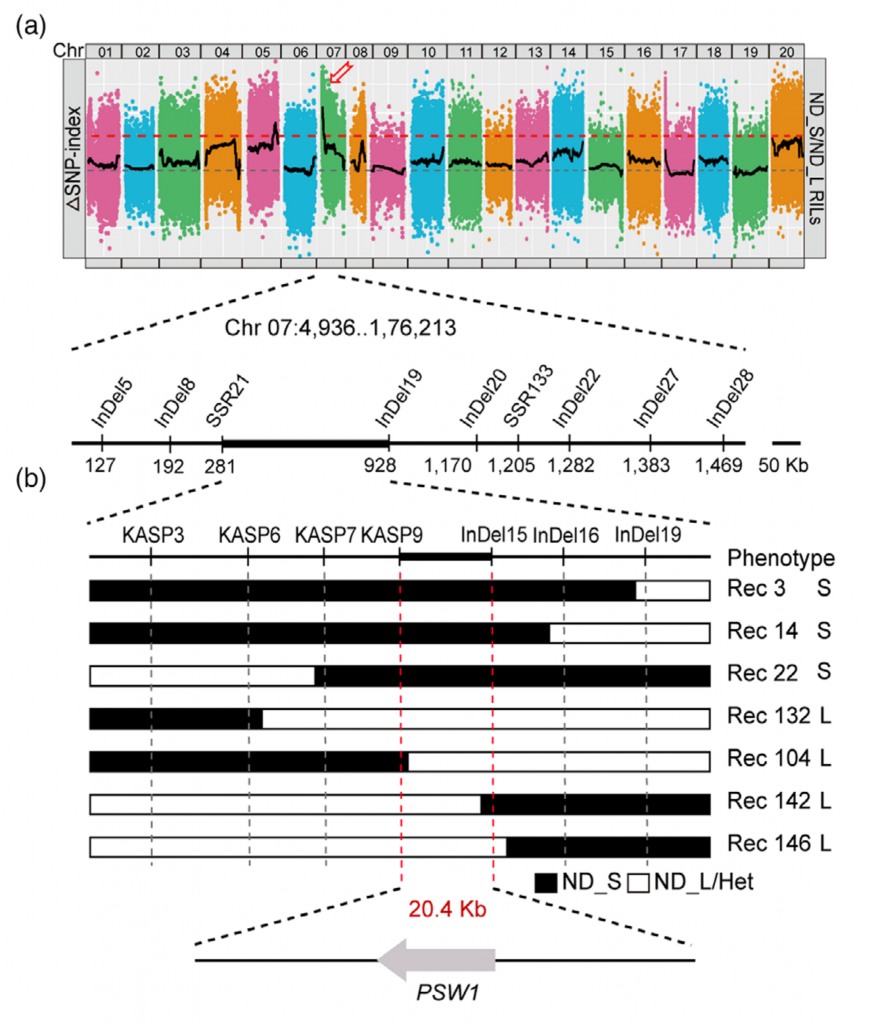
图5-BSA分析
当然,如果想让我们的文章影响因子再上一台阶,兼顾群体的“广度”和“深度”,是更好的选择。
成功案例三

发表期刊:Nature?Genetics
影响因子:30.8
发表单位:浙江省农业科学院等
实验方法:菜用豇豆G98,粮用豇豆G323 基因组Denovo;重测序GWAS:344份全世界收集的豇豆核心种质,其中包括342份栽培豇豆(87份粮用豇豆、244份菜用豇豆和11份未知用途豇豆)和2份野生豇豆;Illumina测序,10x深度;基因单倍型验证:菜用豇豆地方品种 ‘ZN016’ 和菜用豇豆育成品种‘Zhijiang282构建的RIL群体(183 lines)G98和G323构建的F2群体(165 individuals)
百迈客生物为该研究提供了群体测序、基因组测序及部分数据分析服务。
豇豆起源于非洲,在世界范围内作为粮食、蔬菜或牲畜饲料种植。该研究结合PacBio、Hi-C和二代测序,组装了粮用豇豆和菜用豇豆的染色体水平基因组。对包括地方品种、野生品种和育成品种的344个材料进行二代测序,以阐明豇豆基因组的系统进化。
为了研究自然或人工选择对豇豆分化的影响,作者通过选择清除分析比较了三个豇豆亚群之间的基因组选择特征,鉴定出239个与豇豆驯化和改良相关的基因。此外,通过GWAS,挖掘到裂荚性、荚长、单荚粒数、千粒重、可溶性糖、总淀粉和粗蛋白质含量相关基因,并在遗传群体中验证。同时揭示了两个亚种之间基因组结构变异(SVs)的全图谱,为豇豆在全基因组选择下的驯化与改良提供了见解。产量性状和品质性状的差异基因组选择将有助于建立粮用豇豆和菜用豇豆双向改良的遗传资源。

图6-群体选择与GWAS分析
今日分享结束,期待下期精彩内容~~~
]]>
 文章标题:Chromosome-level genome assemblies of two littorinid marine snails indicate genetic basis of intertidal adaptation and ancient karyotype evolved from bilaterian ancestors
文章标题:Chromosome-level genome assemblies of two littorinid marine snails indicate genetic basis of intertidal adaptation and ancient karyotype evolved from bilaterian ancestors
合作单位:中国科学院海洋研究所
发表期刊:GigaScience
影响因子:11.8
研究对象:短滨螺和中华滨螺
百迈客生物为该研究提供了基因组测序服务。
滨螺栖息于岩相潮间带,受到海洋和陆地环境的双重影响,需要根据潮水涨落的节律适应水生和陆生两种完全不同的生境,其广布性、高丰度和对于环境因子胁迫多样且高变动的潮间带环境良好的适应性,使其成为研究环境适应、生物进化和物种形成的潜在模式生物,也是探究生物对于环境压力变化快速响应机制的优良范本,相关的生物学、分类学、系统发育学和生态学研究已经大量开展。此外,细胞遗传学研究结果显示,滨螺具有17条染色体,与两侧对称动物的祖先染色体(ALG)数目一致,因此推演二者之间的核型演化历程具有重要的进化生物学意义。然而由于基因组测序和组装技术的限制,目前滨螺科基因组资源十分匮乏,极大地阻碍了相关研究的顺利开展。
该研究基于三代长读长测序、二代测序和Hi-C测序数据,构建了短滨螺和中华滨螺染色体水平基因组参考序列,并完成了基因结构和功能注释。组装得到两种滨螺基因组大小在900Mb左右,contig N50为3.43Mb和2.31Mb,并成功挂载到17条染色体上,BUSCO评估得分均在93%以上。两个滨螺高质量基因组参考序列作为重要的基因组资源,对于滨螺科及软体动物门系统发育、进化基因组学和生态学等研究都具有十分重要的意义。
通过比较基因组学分析构建了包括两种滨螺在内的11个物种的系统发育树,并估算两种滨螺的分化时间大约在128.22Mya前。进一步的研究结果表明与刺激反应、代谢过程、先天免疫和抗氧化反应相关的基因或基因家族可能在滨螺应对潮间带多种环境因子胁迫的过程中发挥着重要作用,受损核酸和蛋白的修复或降解可能是滨螺在多重压力下抵抗细胞凋亡、维持细胞内稳态的主要方式。相关研究结果为潮间带无脊椎动物适应性进化分子机制的后续研究提供了基础。

图1-11个物种系统发育树
宏观共线性分析结果表明滨螺17条染色体可能由17条ALG经过4次染色体分裂和4次染色体融合演化而来,并推演了由ALG演化到双壳-腹足共同祖先再到滨螺和扇贝的简约演化历程,表明两侧对称动物祖先的基因连锁群在经过上亿年的演化之后在现存物种中仍然得以保留,为未来双壳-腹足共同祖先核型的构建和核型进化速率影响因素的研究提供了参考。

图2-短滨螺、中华滨螺和虾夷扇贝两两间宏观共线性散点图

图3-简约核型演化历程推演及滨螺和扇贝共同祖先(MRCA of PY &Ls)核型构建
内容来源于中国科学院海洋研究所
]]>
文章通过构建本氏烟草端粒到端粒无缺口基因组,对本氏烟草进行了亚基因组分型,进一步确定林烟草(N. sylvestris)和渐狭叶烟草(N. attenuata)最可能是其二倍体祖先物种。研究还深入解析了异源四倍体本氏烟草的着丝粒序列及其表观特征,丰富了我们对本氏烟草基因组进化和着丝粒演化过程的认识。
文章标题:The complete genome assembly of?Nicotiana benthamiana?reveals the genetic and epigenetic landscape of centromeres
合作单位:北京大学现代农业研究院
发表期刊:Nature Plants
研究对象:本氏烟草
百迈客生物为该研究提供了PacBio HiFi、Hi-C、Illumina和RNA-seq测序服务。
研究背景
本氏烟草(Nicotiana benthamiana)是一年生茄科烟草属植物,原产于澳大利亚北部地区,和用于制作香烟的普通烟草(N. tabaccum)是近缘物种。本氏烟草最为人知的是作为植物学和合成生物学研究的模式植物。本氏烟草凭借其对病毒的易感性和在瞬时基因表达的便利性成为了植物科学家的“宠儿”,同时它也是植物天然产物和疫苗异源合成的重要底盘生物。因此,解析本氏烟草的基因组密码对促进植物科学研究和生物制药产业具有重要的价值。本氏烟草是异源四倍体,由两个二倍体祖先在距今500万年-600万年杂交形成,之后基因组演化形成现今的19对染色体。本氏烟草基因组约为2.85Gb,其草图最早发表于2012年,之后的12年间多个改进版本的本氏烟草基因组陆续公布,组装质量有了很大提升,但仍然存在多个缺口与组装注释错误,严重影响了对这一模式生物的功能基因组学的研究进程。
着丝粒是负责细胞分裂过程中染色体平均分配给子细胞的基因组关键区域,也被称为基因组的暗物质区域。因其高度复杂并富含重复序列,着丝粒的序列很难被测序和破译。近年来随着测序技术和生物信息算法的快速发展,包括人类、拟南芥、酵母在内的多个模式生物以及玉米、水稻、辣椒、生菜等作物的着丝粒特征逐渐被揭示。这丰富了我们对这些基因组暗物质的认知,为疾病研究和治疗、作物单倍体育种、人工染色体合成等前沿科学提供理论指导。然而,我们对生物界着丝粒的结构和进化理解仍然处在初期,绝大多数生物的着丝粒区域仍未解析。此外,多倍体生物例如四倍体本氏烟草、四倍体马铃薯、六倍体小麦等,基因组经历了复制、重排和结构变异等事件,在此过程中着丝粒如何演化和维持功能也有待阐明。异源四倍体的本氏烟草为这些问题的解答提供了一个理想的模型。
研究结果
研究团队首先采用单分子测序技术(HiFi,116.7x?+ ONT ultra-long,47.9x),Hi-C(150x)和Bionano(329.6x)光学图谱等多种技术相结合策略,构建了T2T无缺口的本氏烟草基因组(2.85 Gb),实现所有染色体的完整分型组装(图1),并鉴定到所有19个着丝粒和38个端粒,contig N50值达到146.4 Mb。随后的质量评估表明该基因组具有很高的碱基准确性和组装完整性。

图1-本氏烟草T2T基因组全局特征、多倍体进化历史和着丝粒演化进程
研究团队还进一步基于着丝粒特异结合蛋白CENH3的ChIP-seq数据,确定了本氏烟草基因组的完整着丝粒序列,并揭示了其着丝粒全景特征。令人惊讶的是,与辣椒和马铃薯等茄科作物的着丝粒(以LTR/Gypsy反转录转座子为主)不同,本氏烟草着丝粒不仅有Gypsy序列,而且存在大量的卫星(Satellite)DNA的重复阵列,暗示这些着丝粒特异的卫星重复序列是在本氏烟草中新出现的(图2)。经过仔细分析,研究团队证明了本氏烟草着丝粒卫星阵列可能起源于核糖体DNA的基因间间隔序列。
此外,在着丝粒组蛋白CENH3优先占据的区域,Gypsy反转录转座子和核基因组线粒体插入序列(NUMT)广泛侵入本氏烟草着丝粒,表明这些DNA元件在着丝粒功能中起着至关重要的作用。有趣的是,NUMT在本氏烟草着丝粒中的插入具有很强的亚基因组偏好性,并且主要与母体着丝粒周围有关。亚基因组分析表明,卫星阵列的出现可能推动了多倍体后着丝粒的形成(图2)。
最后,该研究提出一个模型来解释本氏烟草着丝粒的进化,即本氏烟草基因组在多倍化后通过新着丝粒形成、卫星序列扩展、反转录转座子的富集和NUMT整合而实现着丝粒进化(图1),丰富了我们对于茄科植物和多倍体植物着丝粒演化的认知。

图2-本氏烟草着丝粒卫星重复序列推动新着丝粒的形成和进化
研究总结
该研究公布了模式植物本氏烟草的T2T无缺口基因组,并揭示了其着丝粒的全景结构及其表观遗传特征,该研究成果具有里程碑意义。本氏烟草完整基因组的破译不但为植物科学研究提供了重要的遗传资源,也将促进本氏烟草作为模式和底盘植物在生物技术领域的广泛应用。
内容来源于北京大学现代农业研究院,侵删
]]>发表期刊:Poultry Science
影响因子:3.8
发表单位:南京农业大学,西藏农牧学院等
研究对象:金陵白鸭
研究方法:全基因组关联分析(GWAS)
百迈客生物为该研究提供了全基因组关联分析(GWAS)测序及部分数据分析服务。

研究背景
鸭子为人类消费提供肉、蛋、羽毛等产品,中国是世界上最大的鸭肉消费国。金陵白鸭做为我国优质的选育品种,具有优良的生长速度和肉质品质。从孵化到上市的整个时期,鸭子的体重逐渐增加,这一过程由复杂的生理和生化机制调控,器官和骨骼的发育是体重增加的关键因素,全基因组关联分析(GWAS)是许多牲畜和家禽物种研究生长和发育的遗传机制的常用方法,然而,现有的一些关于鸭生长发育性状的GWAS研究测序深度较低,发现的候选基因很少。
材料方法
201只雄鸭全基因组测序深度为10×;
GWAS分析:表型:出生体重(BWB)、1周体重(BW1)、3周体重(BW3)、5周体重(BW5)和7周体重(BW7)分析模型:FaST-LMM;EMMAX;LMM;LM 阈值线:log10(p)=5如果一个SNP在2个或更多的模型中被关联,作者认为它是一个与特定性状相关的高可信度SNP。
进化分析:进化树构建;群体结构分析;PCA分析;LD衰减分析等
研究结果
1.金陵白鸭群体体重表型统计
该研究对金陵白鸭(n = 201)在不同时期的体重进行统计,BWB、BW1、BW3、BW5和BW7的体重均值分别为46.93g、127.19g、697.17g、1182.43g和1,888.52g。生长曲线结果显示:金陵白鸭第3周-第5周生长最快,第5周后生长放缓。体重的分散度随着年龄和体重的增加而增加。相邻2周的体重之间存在较强的相关性,BW3与BW1(r = 0.6)、BW5(r = 0.68)和BW7(r = 0.6)显著正相关,BW5与BW7显著正相关(r = 0.82)。但随着时间间隔的延长,性状间的相关性降低,BW1与BW7的相关性不显著(r = 0.21),BWB则与BW3 (r = 0.18), BW5 (r = 0.058),BW7 (r = 0.45)均不显著相关。

图1-表型分析
2.金陵白鸭系统发育、群体遗传结构解析
虽然该研究只有金陵白鸭一个种群,但考虑到繁殖过程中种群分层的潜力,作者进行了系统发育和群体结构分析。系统发育树和PCA结果显示,金陵白鸭可分为5个种群,群体结构表明,当K=6时分群结果最佳,金陵白鸭种群具有丰富的遗传多样性,总SNPs的平均R2为0.24,当R2=0.2,LD的衰减距离约为30kbp。

图2-群体进化分析
3.GWAS分析
该研究对基于测序产生的2,610.50 Gbp的clean data进行分析,Q30为95.55%,样本与参考基因组平均比对率为99.59%,平均覆盖深度为10×,基因组覆盖度为96.51%。作者利用,797,309,337个SNPs,4种关联模型进行后续GWAS分析,其中95个SNP与BWB性状显著相关,这些SNP主要分布在1号染色体和4号染色体上。通过筛选和注释,共检测到5个相关的候选基因:PUS7、FBXO11、FOXN2、MSH6、SLC4A4。针对BW1性状,作者发现了101个与BW1性状显著相关的SNPs,这些SNP主要分布在7号染色体上,共检测到2个与BW1性状相关的候选基因:RAG2和TMEFF2。针对BW3性状,作者发现了112个显著SNPs。这些SNP主要分布在1号染色体和11号染色体上。通过筛选和注释,共检测到4个与BW3性状相关的候选基因:?STARD13、Klotho、ZAR1L和TLE3。同时确定了92个与BW5性状相关SNPs,这些SNP主要分布在1号染色体和2号染色体上,通过注释STARD13、Klotho和ZAR1L与BW5性状相关。此外,作者还鉴定了新的候选基因:KAT2B、KCNH8和SATB1。
BW7性状与33个SNP关联,这些SNP主要分布在1号染色体和2号染色体上。通过筛选和注释,共检测到6个与BW7性状相关的候选基因:PLXNC1、ATP1A1、CD58、FRYL、OCIAD1和OCIAD2。在分析的四个模型的结果,LM识别出了更多的显著位点,曼哈顿图显示出更高的峰值。然而,QQ-plot显示,LM模型中的大部分站点都位于对角线上方,可能具有较高的假阳性。其余3个模型的结果均表现出一致性,而QQ-plot的结果则优于LM模型。

图3-GWAS分析
研究总结
金陵白鸭是一种新开发的品种,因其生长速度快,肉质优良的特点,使其具有重要的经济价值和研究潜力;然而,人们对其体重性状的遗传基础尚不太清楚。该研究对201只金陵白公鸭进行了全基因组重测序,并进行了群体基因组分析,表明金陵白鸭种群具有丰富的遗传多样性。
作者对出生体重(BWB)、1周体重(BW1)、3周体重(BW3)、5周体重(BW5)和7周体重(BW7)进行了全基因组关联分析,4种统计模型比较研究表明,FaST-LMM表现出最优的效率,产生更多的结果和最小的假阳性。
作者发现,PUS7、FBXO11、FOXN2、MSH6和SLC4A4均与BWB相关。RAG2和TMEFF2是BW1的候选基因,STARD13、Klotho、ZAR1L可能是BW3和BW5的候选基因。PLXNC1、ATP1A1、CD58、FRYL、OCIAD1和OCIAD2与BW7相关。这些研究结果为金陵白鸭的选择和育种提供了遗传参考,同时也加深研究者们对白鸭生长发育表型的认识。
]]>期刊名称:Nature Genetics
合作单位:北京大学现代农业研究院
发表时间:2024年7月8日
影响因子:31.7
研究对象:西瓜
测序技术:HIC测序
百迈客生物为该研究提供HIC等建库测序服务。

泛基因组是一个物种中所有个体基因组信息的总和,构建泛基因组可以有效解决单一参考基因组带来的信息缺失和分析偏差。而超级泛基因组则代表一个属内所有物种的基因组信息,尤其蕴含了野生种中丰富的基因组变异,是对泛基因组的进一步扩展,在远缘杂交和基因发掘等方向具有重要应用前景。
西瓜是世界重要的园艺经济作物之一,富含抗氧化剂番茄红素和增强血液循环的瓜氨酸,口味甘甜,享有夏季”水果之王”的美誉,深受全球消费者喜爱。西瓜全球年产量接近1亿吨,我国作为种瓜和吃瓜第一大国,生产和消费了全球总量的60%以上,西瓜产业在乡村振兴和农民致富中占有十分重要的作用。我国西瓜产业的迅速发展离不开优良的品种?,依靠一代代育种家的不懈努力,我国成为世界上西瓜种质创新和品种选育最为活跃的国家,在品种种源方面实现了完全自主可控。随着气候变化的加剧和病虫危害的日益严重,西瓜生产面临着严重的挑战。在现有优良品种的基础上,“植入”缺少的抗病耐逆优良基因,是西瓜种质创新的关键问题,对维持我国西瓜产业高质量绿色发展十分重要。栽培西瓜在驯化和品种改良过程中因为对品质和产量的追求,导致遗传多样性非常狭窄,抗病耐逆基因大量丢失,而在西瓜的祖先即野生西瓜中存在广泛的遗传与表型多样性,具有丰富的抗病耐逆等基因资源,是西瓜遗传改良和种质创新的宝库。
2024年7月8日,北京大学现代农业研究院在国际顶尖期刊《自然-遗传学》上在线发表了题为Telomere-to-telomere?Citrullus?Super-pangenome Provides Direction for Watermelon Breeding的研究成果,是西瓜科研领域的重大突破。
该研究绘制了西瓜属全部7个种的28份代表性材料的端粒到端粒(T2T)高质量基因组图谱,成功构建属级T2T水平超级泛基因组。得到总计768.5Mb,32,513个基因家族的西瓜属泛基因组,是单个西瓜基因组的1.5倍,增加了11,225个栽培西瓜中没有发现的基因。基于T2T高质量基因组图谱,该研究全面比较了西瓜属的着丝粒序列,其丰富的变异和特有的进化关系影响了不同种之间的杂交配对。因此,该超级泛基因组极大地扩展了西瓜遗传改良的基因池。(图1)

图1-西瓜属7个种的28份代表性材料的多样性与泛基因组图谱
利用基因组序列,该研究拓展了西瓜属的分类系统,核实了西瓜起源于非洲的理论依据,并发现栽培西瓜除之前报道的cordophanus亚种外可能还存在其他祖先。另外,我们在西瓜属内发现了三次重大染色体重排事件和两个超长片段倒位。这些染色体重排显著影响了西瓜的抗性、品质相关基因以及三维基因组结构,并在栽培种中得以保留。同时,这些发现也为回交育种过程中避免连锁累赘提供了基因组基础。(图2)

图2-西瓜属进化与栽培西瓜的起源
该超级泛基因组鉴定出西瓜属超过461,987个SV,构建了西瓜图形泛基因组。该研究通过SV-GWAS鉴定了驯化过程中丢失和获得的关键基因,挖掘了与葫芦素含量、含糖量、果肉着色等重要性状相关的功能基因结构变异,发现在西瓜驯化过程中,伴随着多个与甜度增加、果肉变红相关的基因簇扩张,大量抗病功能相关的基因簇丢失。该研究为西瓜育种家提供了较完整的基因组资源,助力深入理解西瓜基因组的复杂性和多样性,从而高效挖掘和利用野生西瓜种中的有利基因。(图3)

图3-西瓜属重要性状基因结构变异图谱
最后,该研究利用野生西瓜的基因组序列和抗病基因信息,通过种间杂交选育形成了抗多种病害的自交系‘PKR6’,并有效确定了西瓜抗枯萎病生理小种候选基因。该研究提供了利用野生种质创制优异育种材料的范例,从而将驯化过程中丢失的抗病基因重新有目的地导入栽培种中改良种质,对加速抗病品种选育、促进西瓜产业高效发展具有深远意义。(图4)

图4-远缘杂交创制优良抗病种质PKR6基因组及枯萎病抗性表型
西瓜属端到端超级泛基因组是较高质量较全面的西瓜属基因组序列图谱和变异图谱,它揭示了西瓜属的基因组演化历史,发掘了西瓜野生种中丰富的遗传多样性,提供了利用野生种质创制优异育种材料的新范例,为其他作物超级泛基因组的构建和野生种质的利用指明了方向。
内容来源于北京大学现代农业研究院,侵删
]]>
文章标题:Assembly of high-quality genomes of the locoweed?Oxytropis ochrocephala?and itsendophyte?Alternaria oxytropis?provides new evidence for their symbiotic rela-tionship and swainsonine biosynthesis
期刊名称:Molecular Ecology Resources
研究物种:黄花棘豆
研究方法:基因组、转录组、重测序、代谢组
百迈客生物为其提供了基因组测序及组装技术服务。
研究背景
黄花棘豆(Oxyvropis ochrocephala Bunge)隶属于豆科棘豆属,为二倍体(2n=16)多年生植物,是我国草原危害最大的毒害草之一。其能够与内生真菌棘豆链格孢菌Alfernaria oxytropis共生,因该内生真菌合成生物碱苦马豆素(swainsonine,SW)而使植株带有毒性,牲畜误食后导致中毒甚至死亡,对畜牧业发展造成了严重影响。
材料与方法
基因组:黄花棘豆;253.39 Gb Illumina+301.02 Gb Nanopore+330.17Gb Hi-C;棘豆链格孢菌;4.40 Gb Illumina+10.09 Gb Nanopore;
转录组:黄生棘豆无菌幼苗、黄生棘豆及其内生菌共生苗、棘豆链格孢菌;每个材料3个生物学重复;
重测序:从中国3个省13个地理位置采集的黄花棘豆中分离的41株棘豆链格孢菌;19x:代谢组:LC-MS;
代谢组:41株棘豆链格孢菌苦马豆素含量测定
研究结果
1.黄花棘豆及A.oxytropis基因组组装注释
作者利用三代Nanopore、二代Illumina测序技术以及Hi-C技术测序组装获得一个高质量染色体水平的黄花棘豆基因组:其基因组大小为930.94 Mb,contig N50为1.40 Mb,scaffold N50为121.79 Mb,共注释到31,700个蛋白质编码基因。利用三代Nanopore和二代Illumina测序技术测序组装获得一个高质量的棘豆链格孢菌基因组:其基因组大小为74.48 Mb,contig N50为8.87Mb,共注释到10,657个蛋白质编码基因。
2.黄花棘豆及A.oxytropis基因组进化和全基因组复制分析
黄花棘豆基因组进化分析显示,其大约于42.62百万年前分化出来,且与蒺藜苜蓿、大豆具有较近的亲缘关系。黄花棘豆与蒺藜苜蓿之间基因的共线性关系更好,但染色体发生了部分片段的重排,这些重排的基因主要参与代谢途径。黄花棘豆基因组中6,938个基因家族发生收缩,1,862个基因家族发生扩张,在进化过程中发生了一次WGD事件。A.oxytropis基因组进化分析显示,A.oxytropis与同属的其他真菌聚在一起,与不同属中产苦马豆素的真菌相比,A.oxytropis具有该物种最丰富的特有基因家族,主要与代谢途径相关。基于SW合成基因簇的关键基因swnK的系统进化分析发现,有14个目的真菌均含有该基因簇。基于ITS系统进化分析发现,Altemaria属内产苦马豆素(含有swnK基因)的物种聚在一起,与不含swnK基因的物种分离。

图1.黄花棘豆与其他10个物种的比较基因组学分析
3.黄花棘豆与A.oxytropis基因表达
黄花棘豆与A.oxytropis共生/非共生状态下的转录组分析显示,对寄主而言,其与内生真菌共生后差异基因主要与防御以及次级代谢相关;对内生真菌而言,其在寄主体内共生时,参与脂肪酸代谢、氮代谢以及降解细胞壁相关的差异基因被大量诱导上调表达,同时共生诱导了大量SW合成基因的上调表达。
4.A.oxytropis遗传变异影响苦马豆素含量
该研究丰富了前期推测的SW合成通路,进一步分析显示,一个编码P5CR的基因位于SWN簇上,也包含在SWN的全长序列中。对从中国3个省13个地理位置采集的黄花棘豆种分离的41株A.oxytropis的重测序分析显示,一个位点SNP19996与SW含量相关。

图2.SW基因表达、基因结构以及SWN基因簇中SNP多样性、A.oxytropis体内SW含量
研究总结
作者构建了高质量黄花棘豆基因组(958.83Mb),及其共生真菌A.oxytropis基因组(74.48Mb,contig N508.87Mb),并对SW生物合成基因进行了细化。41株A.oxytropis的重测序分析找到与SW含量相关位点,转录组分析发现了与宿主的防御和次级代谢相关的差异表达基因(DEGs)。在内生菌中,DEGs与细胞壁降解、脂肪酸和氮代谢有关。共生关系诱导了大部分SW生物合成基因的上调。
]]>
文章标题:Chromosome-scale genome assembly clarifies the mechanism of flooding tolerance and evolutionary?history of?Myricaria laxiflora
期刊名称:Industrial Crops & Products
合作单位:西南大学、重庆市林业科学研究院、重庆城市管理职业学院
研究物种:疏花水柏枝
研究方法:基因组学
百迈客生物为该研究提供了基因组测序和部分分析工作。
疏花水柏枝(Myricaria laxiflora),柽柳科,是长江流域天然存在的宝贵植物资源,对淹没的栖息地具有较好的适应性,每年可以忍受大约五个月的淹没。洪水对植物的生长、发育和产量产生负面影响,严重时可能导致死亡。全球气候变化使洪水更加常见和严重。研究疏花水柏枝对极端洪涝环境的适应机制,寻找关键功能基因,为种质改良提供理论和遗传依据。
本研究借助三代PacBio HiFi和Hi-C等技术,构建了高质量染色体水平的疏花水柏枝基因组,最终组装大小为1.34Gb,contig N50=11.69Mb。二代、三代回比比对率分别为94.14%和99.75%,CEGMA和BUSCO评估分别为93.67%和96.47%,表明组装的基因组完整性比较高。结合转录组预测、同源预测和从头预测的方式共得到23,719个蛋白质编码基因。

图1-疏花水柏枝表型和基因组特征
比较基因组研究发现疏花水柏枝与苦荞(F. tataricum)的亲缘关系最为密切,大约在65-81百万年前(Mya)发生分化;在19.74 ~ 24.60 Mya之间发生了一次全基因组复制(WGD), 4dTV分析证实了这一点。在0.15 Mya左右存在一个长末端重复(LTR)插入。这些基因组变化可能与疏花水柏枝在进化过程中所经历的环境剧变(如青藏高原隆升)有关。基因家族分析显示疏花水柏枝的特有基因家族与能量代谢和信号转导密切相关;85个基因在疏花水柏枝基因组中经历了正选择,这些基因在与氨基酸合成和代谢相关的KEGG通路中显著富集。

图2-疏花水柏枝系统进化研究
作者通过比较水淹和对照条件下疏花水柏枝形态、生理和转录组学的变化,分析了植物对水淹的适应机制。总体而言,在完全淹没的状态下,疏花水柏枝采用“静止”的策略。具体来说,在淹水条件下,光合作用减弱,生长停滞,糖酵解被激活,抗氧化酶系统增强。乙烯反应途径可能是这一过程的调控因子,MlERF-VII基因(Mla11G026380)可能是调控这一过程的关键基因。

图3-疏花水柏枝水淹胁迫响应信号通路
小 结
本研究成功构建了一个染色体水平的疏花水柏枝基因组,并对其特征进行了分析。进一步结合形态学、生理学和比较转录组学分析评估了该物种适应极端洪水环境(完全淹没)的遗传机制。本研究对疏花水柏枝种质资源的保护和资源的开发利用具有重要意义。
]]>