



在动植物遗传育种中,通过连锁关联分析对数量性状位点(Quantitative trait locus,QTL)和质量性状基因进行定位,对于加速育种改良的进程具有重要的意义。对于植物群体,通常利用性状具有显著差异的两个亲本进行杂交,再通过自交、回交或者其他方式来构建家系群体。家系群体按照遗传稳定性划分可分为暂时性分离群体和永久性分离群体,连锁分析是基于家系群体利用连锁的原理研究相关基因与遗传标记的关系的一种研究方法。全基因组关联研究(GWAS)是一种在群体水平上研究基因型与表型之间关联性的分析方法,更适用于自然群体基因功能定位。
以下小编列举三篇成功案例,解析群体在QTL定位中的应用。
当我们针对一群体,关注性状较多时,可参照案例一,整篇文章通过群体图谱构建,QTL定位后几乎没有验证工作,可以帮我们拿到一个群体QTL的基础数据。
案例一

发表期刊:Plant Physiology and Biochemistry
影响因子:6.1
合作单位:江苏省徐淮区徐州农业科学研究所农业农村部甘薯生物学与遗传改良重点实验室
实验方法:Xin24×Yushu10杂交中选择212个F1材料: 特异性位点扩增片段测序(SLAF-seq);遗传图谱构建;数量性状位点(QTL)定位;GWAS关联(F1群体)
表型检测:多年多表型收集
百迈客生物为该研究提供了群体测序及部分数据分析服务。
甘薯作为一个全球重要的作物,其含有丰富的营养成分以及对不同环境的适应能力。然而,由于其自交不亲和,高杂合度等特性,使其遗传特征的研究相对较少。作者从Xin24×Yushu10杂交中选择212个F1材料,SLAF-seq测序,获得亲本26.73×,子代52.25×的测序数据,依据SNP及百迈客生物自主研发的HighMap构图软件,生成一个长度为2441.56 cM、平均图距为0.51 cM的遗传图谱。
基于连锁图谱,鉴定出26个QTL,解释了6.3-10%的表型变异,包括6个最长藤蔓长度 QTL、6个单株产量 QTL、10个干物质含量QTL、1个淀粉含量 QTL、一个可溶性糖含量QTL和2个类胡萝卜素含量QTL。该研究结果对甘薯的标记辅助育种和基因克隆具有重要意义。
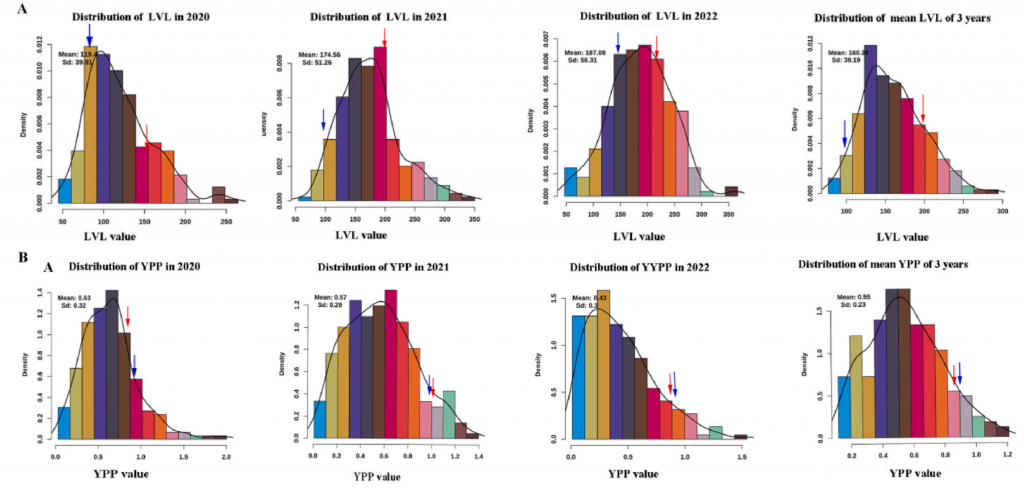
图1-表型检测
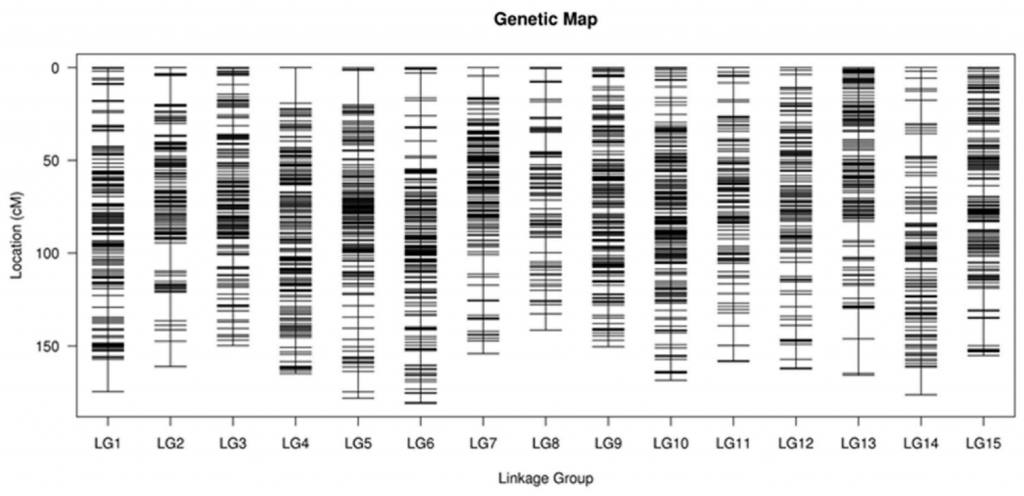
图2-遗传图谱构建
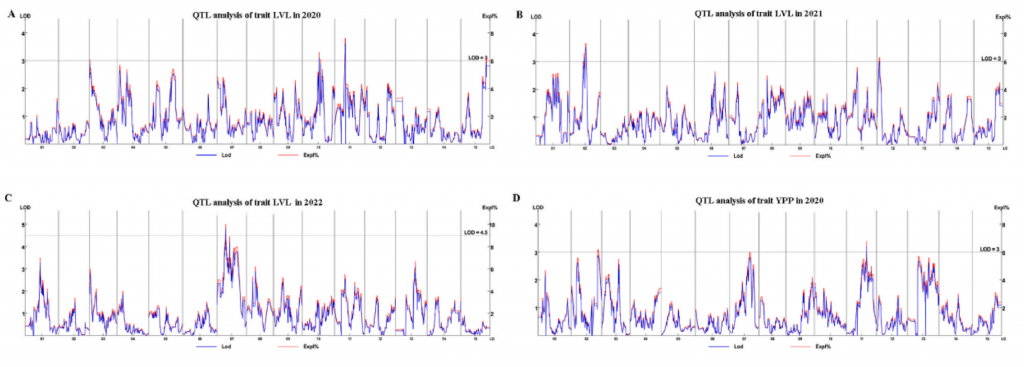
图3-QTL定位
前文是对多个性状的连锁分析,当我们关注单个性状时,BSA无疑是高性价比的初定位选择,当然,这就意味着我们得做到基因的精细定位与克隆,除去传统图位克隆的方式,转录组,蛋白组,自然群体GWAS,基因组都可助力基因克隆,如果想发高水平的文章,基因的功能探索也是必不可少的。
成功案例二

发表期刊:Plant Biotechnology Journal
影响因子:10.1
发表单位:河南农业大学
实验方法:荚果大小/重量差异显著2份Virginia-type花生材料( ND _ L和ND _ S)杂交,产生遗传群体;F2:3群体BSA-seq(20+20混池);F6:7 和F6:8群体精细定位;基因克隆;系统进化分析;亚细胞定位;免疫荧光;酵母双杂交;pull-down;CO-IP;番茄拟南芥转化实验等。
百迈客生物为该研究提供了群体测序及部分数据分析服务。
花生荚果大小是决定花生产量的关键农艺性状,为了鉴定控制花生荚果大小的基因,该研究对188份核心种质进行鉴定,并选取荚果大小/重量差异显著的2份花生材料构建F2群体,BSA-Seq获得了288.58 Gb的原始数据。利用285914个高质量SNPs 和70 759个 InDel,将控制荚果大小的基因定位在07染色体1.17 Mb的区间内。作者从F6:7和F6:8群体中开发了15个多态性标记并对个体进行基因分型,精细定位QTL到KASP 9 和In Del15之间20.4 kb的区间内。该区间仅包含1个预测的非同义突变基因和InDels,将该基因命名为PSW1。
PSW1编码一个LRR – RLK蛋白激酶,等位基因PSW1HapII赋予了PSW1更高的表达水平和对其辅助受体AhBAK1更强的亲和力,以上调PSW1 – based途径,调节花生荚果大小。此外,PSW1HapII的过表达增加了多种植物的种子/果实大小。
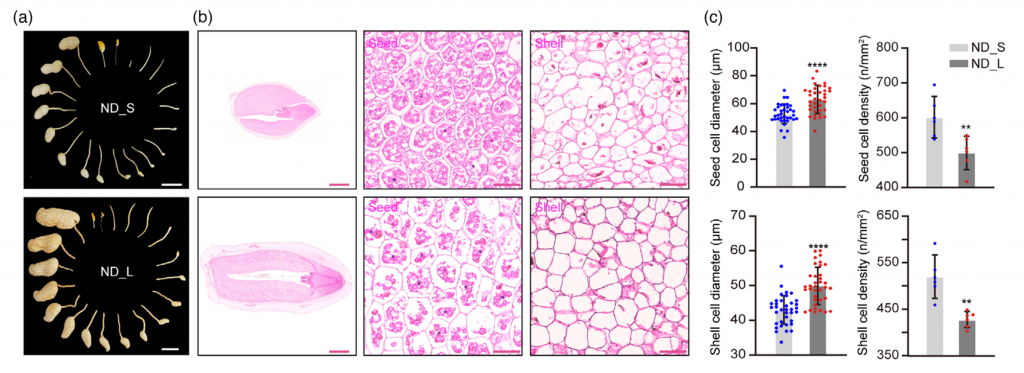
图4-表型检测
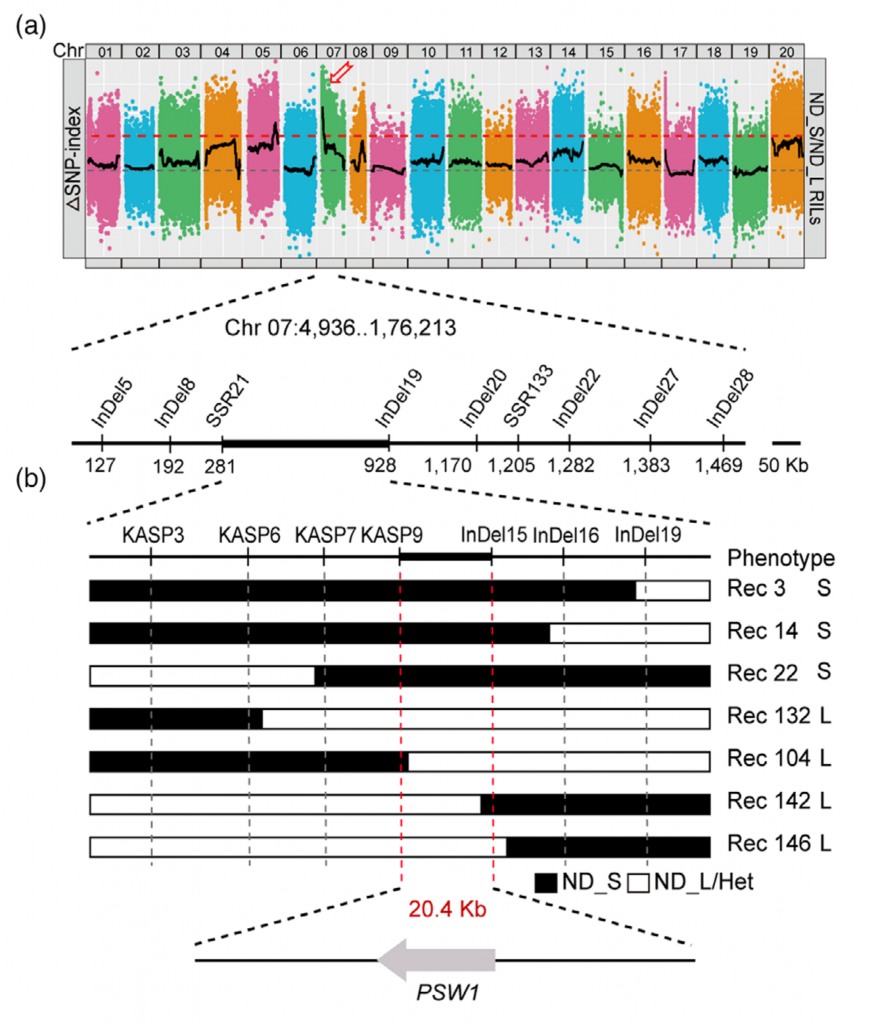
图5-BSA分析
当然,如果想让我们的文章影响因子再上一台阶,兼顾群体的“广度”和“深度”,是更好的选择。
成功案例三

发表期刊:Nature?Genetics
影响因子:30.8
发表单位:浙江省农业科学院等
实验方法:菜用豇豆G98,粮用豇豆G323 基因组Denovo;重测序GWAS:344份全世界收集的豇豆核心种质,其中包括342份栽培豇豆(87份粮用豇豆、244份菜用豇豆和11份未知用途豇豆)和2份野生豇豆;Illumina测序,10x深度;基因单倍型验证:菜用豇豆地方品种 ‘ZN016’ 和菜用豇豆育成品种‘Zhijiang282构建的RIL群体(183 lines)G98和G323构建的F2群体(165 individuals)
百迈客生物为该研究提供了群体测序、基因组测序及部分数据分析服务。
豇豆起源于非洲,在世界范围内作为粮食、蔬菜或牲畜饲料种植。该研究结合PacBio、Hi-C和二代测序,组装了粮用豇豆和菜用豇豆的染色体水平基因组。对包括地方品种、野生品种和育成品种的344个材料进行二代测序,以阐明豇豆基因组的系统进化。
为了研究自然或人工选择对豇豆分化的影响,作者通过选择清除分析比较了三个豇豆亚群之间的基因组选择特征,鉴定出239个与豇豆驯化和改良相关的基因。此外,通过GWAS,挖掘到裂荚性、荚长、单荚粒数、千粒重、可溶性糖、总淀粉和粗蛋白质含量相关基因,并在遗传群体中验证。同时揭示了两个亚种之间基因组结构变异(SVs)的全图谱,为豇豆在全基因组选择下的驯化与改良提供了见解。产量性状和品质性状的差异基因组选择将有助于建立粮用豇豆和菜用豇豆双向改良的遗传资源。

图6-群体选择与GWAS分析
今日分享结束,期待下期精彩内容~~~




 京公网安备 11011302003368号
京公网安备 11011302003368号